
Last modified on 29 December 2006.
Introductory Statements:
Our objectives are to predict RNA secondary and tertiary structure from its underlying sequence. Given the very large number of theoretically possible secondary structure models (discussed in more detail below) for an RNA sequence, our limited expertise on the thermodynamic, kinetic, and other factors that influence the folding of RNA molecules, and our rudimentary knowledge about RNA structural motifs, we are currently unable to accurately and reliably predict the correct secondary structure from a single RNA sequence (a partial list of references is available). However, it is possible to accurately predict secondary and even some tertiary structure basepairings from nucleotide sequence information.
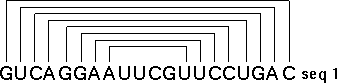
The method that achieves this goal is "comparative sequence analysis" and it is based on this very simple principle: a single RNA secondary and tertiary structure can be formed from different RNA sequences. At the present time, our sequence analysis-based secondary and tertiary structure models are composed primarily of (base) paired and unpaired nucleotides. Basepairs are usually G:C or A:U, and are adjacent and antiparallel with one another to form helices. Since a helix can be formed from two sets of sequences that are not identical with one another, we search for positions (in an alignment of homologous sequences) that vary coordinately (covary) to maintain Watson-Crick pairing within a potential secondary structure helix. The search for coordinated base substitutions in an alignment of homologous sequences is called covariation analysis. Note in the example below, Sequence #1 forms seven consecutive basepairs that are arranged antiparallel with one another. Sequence #2 also forms a helix of the same length with A:U and G:C basepairs.
 |
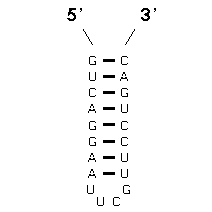 |
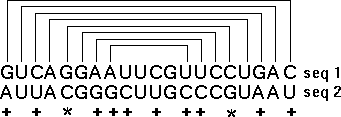 |
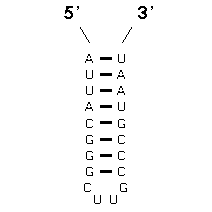 |
Note that several compensatory base changes occur between Sequences #1 and #2. Of the 20 homologous nucleotides in sequences 1 and 2, 12 (or 60%) are different. Of these 12 nucleotides, 10 are associated with basepairs. The essence of covariation analysis is the identification of positions that have the same or similar patterns of variation in an alignment of sequences. Base pairs are then inferred from the strongest or best sets of covarying positions.
The comparative method was validated when the crystal structure of tRNA became available in 1976, when all of the secondary structure interactions predicted by this sequence analysis were present in the crystal structure solution (references). As a result of this success, other RNA molecules have been analyzed with a sequence covariation approach. These include the 5S, 16S, and 23S rRNAs, as well as group I introns, U-RNA, ribonuclease (RNase) P, the 7S signal recognition particle RNA, and telomerase RNA (reviewed in: Gutell R.R. (1993). Comparative Studies of RNA: Inferring Higher-Order Structure from Patterns of Sequence Variation. Current Opinion in Structural Biology 3:313-322.).
Early in the development of this analysis, a helix was considered phylogenetically proven when at least two compensatory base changes were present within a potential helix (Woese C.R., et al. (1980). Secondary structure model for bacterial 16S ribosomal RNA: phylogenetic, enzymatic and chemical evidence. Nucleic Acids Research 8(10):2275-2293). After the initial secondary structure models were proposed, each new RNA sequence was compared to its existing model to test the validity of the base pairs in the model and to determine new secondary and tertiary structure interactions. Thus, the comparatively inferred secondary and tertiary structure models have been continually refined and substantiated as the number of sequences has increased significantly and the covariation methods were improved.
Paradigms and Algorithms: Within this Structure Prediction With Comparative Sequence Analysis section, we present a historical perspective of the covariation methods, starting with the early visualization of Watson-Crick covariations within potential helices [Visual Methods], progressing to simple number pattern computational searches that are independent of structural constraints [The Number Pattern Method], and continuing with chi-square based statistics [Chi-Square Statistical Algorithms], phylogenetic event counting [Phylogenetic Algorithms], and searches for only Watson-Crick and Watson-Crick/GU basepairs and covariation purity scores [Empirical Algorithms]. We conclude the covariation methods section with our current "collective scoring" criteria [Collective Scoring], and then apply it to several RNA molecules, including tRNA, and 5S, 16S, and 23S rRNA.
Structure Definitions: This section presents our definitions of terms used to describe RNA structure and examples of these structural elements, to complement the analysis provided elsewhere in this site.
Reference Secondary Structure Model Diagrams: This section contains diagrams of the current secondary structure models for the reference sequence for each molecule (e.g., Saccharomyces cerevisiae Phe for tRNA). Model diagrams are presented in two forms (shwoing either individual nucleotides or schematics to represent the primary structure) and available for download in two file formats (PostScript and PDF). Also available from this page are circular and histogram diagrams, which provide an alternative view of the secondary structure, and additional diagrams with specific purposes (for example, a diagram showing our tentatively proposed interactions or the changes since the last published version of the model).
Our last comparative analysis resulted in numerous refinements in our 16S and 23S rRNA secondary and tertiary structure models, which are presented here. In our nucleotide frequency tables (under Section 2A), we present the actual comparative data supporting every basepair and base triple in tRNA, 5S, 16S, and 23S rRNA.
Note: Since we are largely ignorant to the types of structural conformations that unpaired nucleotides can have, our secondary and tertiary structure models are essentially composed from base pairs and their arrangements with one another. Unpaired nucleotides (except for a few base triples) currently are not structured in our comparatively inferred RNA structure models.
For the interested reader, a partial (and biased) list of review articles is available.
Number of Possible Secondary Structure Models
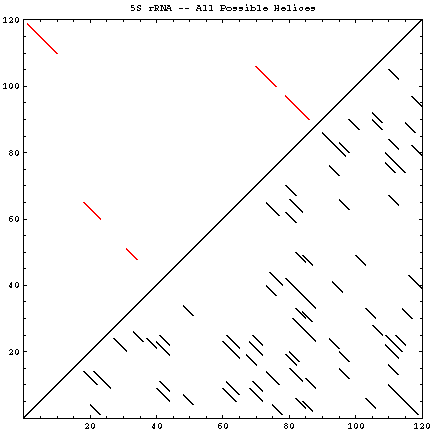
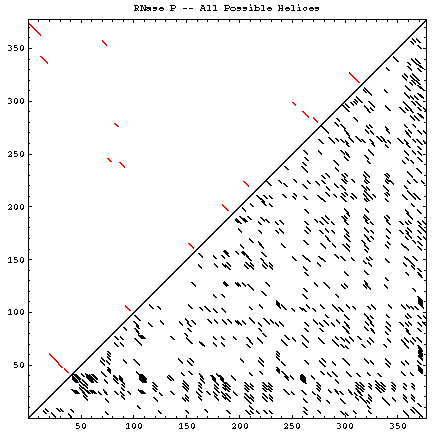
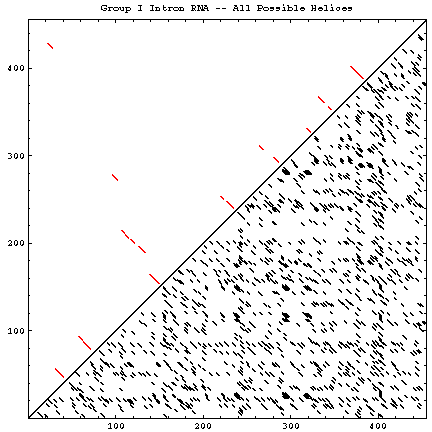
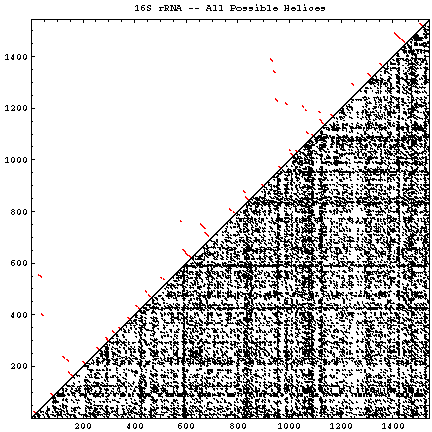
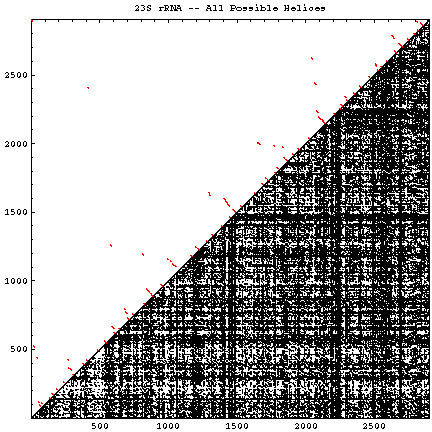
A single RNA sequence can be folded into a large number of theoretically possible secondary and tertiary structures. The number of possible secondary structure models has been estimated by Zuker and Sankoff (references) to be greater than 1.8n, where n is the number of nucleotides in a given sequence [assuming that all four nucleotides (A, C, G, and U) occur with equal probability and at random in the RNA sequence].
The figure below helps to visualize the scale of this problem. Below and to the right of the diagonal are shown all of the possible helices of length 4 or greater, with primarily Watson-Crick composition. Above and to the left of the diagonal are the helices found in the experimental and comparative structures.
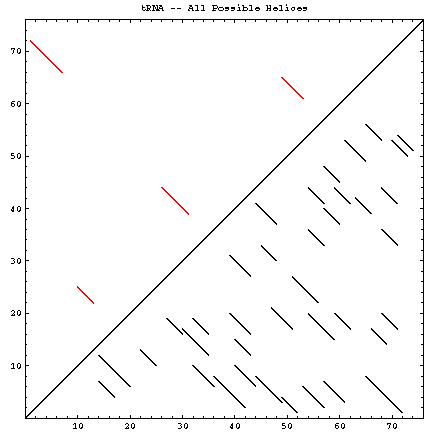
| Molecule (Method) | Reference Sequence / GenBank Accession # | Length | # of Helices (P) | # of Secondary Structures (Z) | # of Helices (CM) | Figures |
|---|---|---|---|---|---|---|
| tRNA (X) | Saccharomyces cerevisiae (Phe) / K01553 | 76 | 37 | 2.5 * 1019 | 4 | GIF | PS | PDF |
| 5S rRNA (C, X) | Escherichia coli / D12500 | 120 | 71 | 4.3 * 1030 | 5 | GIF | PS | PDF |
| RNase P (C) | Escherichia coli | 377 | 784 | 1.7 * 1096 | 17 | GIF | PS | PDF |
| Group I Intron (C, X) | Tetrahymena thermophila / V01416 | 414 | 1307 | 4.8 * 10105 | 19 | GIF | PS | PDF |
| 16S rRNA (C) | Escherichia coli / J01695 | 1542 | 14684 | 4.3 * 10393 | 58 | GIF | PS | PDF |
| 23S rRNA (C) | Escherichia coli / U00006 | 2902 | 51442 | 6.3 * 10740 | 105 | GIF | PS | PDF |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}