
Last modified on 10 January 2000.
(mid-1980's-early 1990's) Our First Comparative Algorithm: The Number Pattern Method
As the sequence databases grew, new comparative methods were developed to handle the added information. The number pattern method, the first major development, ordered attempts to determine more structure. Each position in a sequence alignment was converted to a number pattern, which showed how much or how little the position varied. Similar patterns could then be sorted together, providing the opportunity to reevaluate the secondary structure model and to detect new interactions.
To construct the number pattern, a standard alignment, which contains individual sequences as horizontal lines, is rotated ninety degrees so that each horizontal line represents a single position in each sequence and that a single sequence is now a column, as shown in this segment from tRNA:
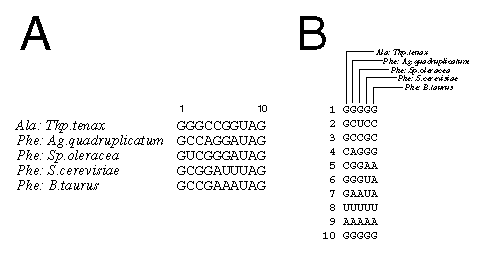 |
The nucleotides are then transformed into the number pattern using these criteria:
- Non-ACGU positions are assigned "0."
- The first nucleotide (ACGU) found and all subsequent occurences of that nucleotide at that position is 1.
- The transition nucleotide is 2.
- The first transversion nucleotide to appear is assigned 3.
- The other transversion nucleotide is 4.
In the example above, the pattern for position 1 would be 11111, since the guanosine is completely conserved. Position 2 has the pattern 13433; since A (transition from G) does not appear, the number 2 does not appear in the pattern. The pattern for position 6 is 11132, since U is a transversion and A a transition from G.
The resulting numerical strings can be sorted (by computer) to group like sequences together. Two positions having identical pattern are likely to interact when they are the only positions having that pattern. As the length of the molecule increases, the probability that identical patterns will occur decreases. Secondary structure interactions tend to have more complex number patterns.
In the table below, partial results of the number pattern method are shown for the tRNA-5 alignment. (You may view the complete results in a separate window.) First is shown the number pattern as generated. In the right-hand cell is the sorted number pattern. For both sets of data, the first column (#) contains the position number. The second and third columns are the number pattern (numpt) and nucleotide composition (nucpt), respectively. The fourth column (pairing) indicates the pairing partner (bp-xx) for each paired position and how well the method identifies the base pair. If "BP" appears, the pair is unambiguously identified. An asterisk (*) at the end of the entry shows that the position and its partner share a number pattern but that pattern is not unique.
| Sorted by Position Number | Sorted by Number Pattern |
|---|---|
# numpt nucpt pairing -- ----- ----- ------- 1 11111 GGGGG bp-72* 4 13444 CAGGG bp-69 6 11132 GGGUA BP-67 8 11111 UUUUU bp-14* 10 11111 GGGGG bp-25* 11 12222 UCCCC bp-24* 12 10222 CNUUU bp-23 13 12222 UCCCC bp-22 14 11111 AAAAA bp-8* 15 11111 GGGGG bp-48* 22 11111 GGGGG bp-13 23 12222 GAAAA bp-12 24 12222 AGGGG bp-11* 25 11111 CCCCC bp-10* 26 12211 GAAGG BP-44 35 12222 GAAAA 37 12212 GAAGA 44 12211 AGGAA BP-26 46 12222 AGGGG 48 11111 CCCCC bp-15* 50 13121 CGCUC BP-64 52 11231 GGAUG BP-62 57 12212 GAAGA 62 11231 CCUAC BP-52 64 13121 GCGAG BP-50 69 13443 GCUUC bp-4 72 11111 CCCCC bp-1* |
# numpt nucpt pairing -- ----- ----- ------- 12 10222 CNUUU bp-23 1 11111 GGGGG bp-72* 8 11111 UUUUU bp-14* 10 11111 GGGGG bp-25* 14 11111 AAAAA bp-8* 15 11111 GGGGG bp-48* 22 11111 GGGGG bp-13 25 11111 CCCCC bp-10* 48 11111 CCCCC bp-15* 72 11111 CCCCC bp-1* 6 11132 GGGUA BP-67 67 11132 CCCAU BP-6 52 11231 GGAUG BP-62 62 11231 CCUAC BP-52 26 12211 GAAGG BP-44 44 12211 AGGAA BP-26 37 12212 GAAGA 57 12212 GAAGA 11 12222 UCCCC bp-24* 13 12222 UCCCC bp-22 23 12222 GAAAA bp-12 24 12222 AGGGG bp-11* 35 12222 GAAAA 46 12222 AGGGG 50 13121 CGCUC BP-64 64 13121 GCGAG BP-50 69 13443 GCUUC bp-4 4 13444 CAGGG bp-69 |
Some interesting cases are shown in this excerpt:
- Four base pairs (6:67, 26:44, 50:64, and 52:62) are unambiguously detected; no other positions have the same number pattern.
- 4:69 and 12:23 are not cleanly detected due to differences in the pattern at one place; positions 4 and 69 occur in close proximity in the sorted pattern and might be recognized as pairing partners, but 12 and 23 are further separated and are less likely to be noticed. (Note also that if position 12 had the pattern "12222," the 12:23 pair could not be unambiguously assigned.)
- Two unpaired positions, 37 and 57, share a number pattern and present a possible false positive signal.
- Invariant positions all have a number pattern consisting only of 1's; although many sets of pairing partners appear in this group, these base pairs cannot be unambiguously assigned.
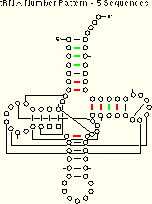
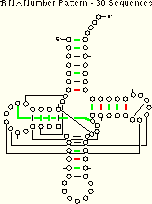
Enlarging the sequence database affects the efficiency of the number pattern method. Consider the comparison of the results from the tRNA-5 (five sequences) and tRNA-30 (thirty sequences) alignments:
 |
 |
| tRNA-5 Number Pattern (Five tRNA Sequences) 6 (10) of 27 Pairs Identified |
tRNA-30 Number Pattern (Thirty tRNA Sequences) 4 (15) of 27 Pairs Identified |
| tRNA-5 Complete Results | tRNA-30 Complete Results |
| Composite Figure for this table. | tRNA Secondary Structure Diagram (PostScript) |
More interactions are implied using the larger alignment, but in this case fewer are unambiguous. The amount of ambiguity is related to the similarity of the sequences involved.
| Alignment | Low % Similarity | High % Similarity |
|---|---|---|
| 5 tRNA Sequences | 41.6% | 85.1% |
| 30 tRNA Sequences | 30.5% | 85.1% |
| 895 tRNA Sequences | 23.6% | 100.0% |
The number pattern method has the drawback that correlating pairs are missed when the number pattern at the two positions is not strictly identical. This can happen when one position undergoes single base changes, as in the tRNA 4:69 base pair discussed above. Variations involving unorthodox base pairs in helical regions are often missed because of irregularities introduced into the number pattern by the unorthodox pairs.
Despite these drawbacks, the number pattern method represents an advance in comparative techniques, since two of the biases that affected the visual pattern recognition methods do not interfere with the number pattern method. First, this method is equally able to find Watson-Crick and noncanonical interactions. Only the number pattern is important for the comparative analysis; the identities of the bases do not play a role in this step. Therefore, the number pattern method allowed more possible pairings, increasing the scope of the search and determining a greater portion of a molecule's secondary structure. In addition, the constraint of structural context was relaxed; this method is context-independent, and is not potentially hampered by visual bias.
The consequence of context-independence is that while many secondary structural interactions were detected, the method also identified interactions which occurred in isolation from other pairings. This ability enabled the search for tertiary interactions to begin in earnest as the sequence databases continued to grow, utilizing the more sophisticated pattern matching technique. In 1985, the following criteria were proposed for identifying tertiaries using number pattern data:
- Two positions occur in mutual proximity in the primary or secondary structure;
- Clusters of positions in two different regions of the structure maintain coordinated patterns of variation;
- Experimental observations strongly support interactions;
- Patterns show differences within, rather than between, phylogenetic groups.
In summary, the successes of the number pattern method include:
- Confirming many proposed base pairs in E. coli 16S and 23S rRNA, while maintaining the general secondary structure;
- Implying that alternative, mutually exclusive helices (helical 'switches') were unlikely to exist in 16S rRNA.
- Uncovering basic principles of RNA structure:
- Most interactions detected are canonical (Watson-Crick and GU) pairings, despite the fact that these are not selected for by the method itself.
- Canonical pairings occur in contiguous, antiparallel arrangements.
- Covariance between any two positions is independent of the pattern of change at flanking positions.
- The majority of covarying positions are flanked by other correlating positions and arranged in a helical fashion.
- Most importantly, the first tertiary interactions were proposed for 16S rRNA.